

Highlight
Sequencing by Ligation Variation with Endonuclease V, Deoxyinosine, and SAWTooth The Sequencing Analysis Workbench Tool
Sequencing by Ligation
Achievement/Results
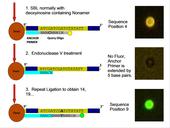
Sequencing-by-ligation (SBL) is one of several next-generation sequencing methods that have been developed for massive sequencing of DNA immobilized on arrayed beads (or other clonal amplicons). Polony Sequencing utilizes a fixed bead array and SBL to obtain DNA sequence information. Biotinylated template DNA is isolated in a PCR solution and added to streptavidin-coated beads. Through a process of emulsion PCR, the beads become clonally covered with multiple copies of a single template strand. In SBL, an anchor primer is hybridized to a known region of the template DNA, usually a linker or adaptor that has been ligated onto a fragment of an unknown DNA sequence. Then, using a series of degenerate query oligos that have fluorophores, the DNA adjacent to the known region is sequenced through a series of hybridizing an anchor primer, ligation of a query oligo, then denaturing the DNA and clearing all signal and repeating.
Images captured in four fluorescent channels, one corresponding to each base pair, serve as data. On every frame, every bead’s coordinates are recorded, as well as the signal the bead gave for a base pair position. After multiple cycles of biochemistry and imaging, a sequence is generated for every bead, which is then used as the raw sequence for alignment. SBL has the advantage of being easy to implement and accessible to all, because it can be performed with off-the-shelf reagents. However, SBL has the limitation of very short read lengths. To overcome the read length limitation, complex library preparation processes have been developed, which can be time-consuming, difficult, and result in low complexity libraries. Antoine Ho, an INCBN IGERT Graduate Trainee, is working in the laboratory of his main advisor, Prof. Jeremy Edwards from the Dept. of Molecular Genetics and Microbiology at UNM School of Medicine on a variation of traditional SBL protocols called cyclic SBL (cSBL), which extends the number of sequential bases that can be sequenced by using Endonuclease V to recognize an incorporated deoxyinosine site that serves as a query primer and to clip the DNA, thus leaving a ligatable end extended into the unknown sequence for further SBL cycles. Virtually all next-generation sequencing platforms generate gigabytes of data per run, often in the form of mate-paired (or single sequence) short-reads. This requires the analysis and mapping of several billion mate-paired reads when used for whole-genome sequencing. An efficient algorithm to perform this mapping to a large reference genome, itself comprising several giga-base-pairs, is essential, given the very large dataset sizes.
With the assistance from his co-advisor, Prof. Susan Atlas from the Dept. of Physics, Antoine Ho and his colleagues have developed in-house a dedicated software package called SAWTooth (Sequencing Analysis Workbench Tool), whose core functionality is the efficient mapping of short-read sequencing data to a reference genome, outperforming other popular codes used in genome alignment by ~100-fold or more. SAWTooth also implements several ancillary applications for validation and statistical analysis of mapping results. All fast contemporary mapping algorithms rely on indices. These auxiliary data structures facilitate mapping sequences to a reference genome. These indices generally fall into two broad categories – suffix trees and hash indices. Traditionally, the construction and use of suffix-trees imposed prohibitive memory requirements, though in recent years, innovations in the field of compressed text indices have rendered suffix-based methods feasible for whole-genome indexing, though still not optimal. SAWTooth utilizes hash indexing, a well-known referencing data structure, which allows key-based data retrieval in constant, O(1) time, making it the fastest of all data retrieval structures. In principle, there are some limitations of general hash indices that may limit their performance or impair their usefulness. Keys are not ordered, so sorted lists and range searches are not intrinsic operations on the data structures. Also, hash function may generate the same hash for multiple keys, resulting in a collision. Resolving collisions requires extra processing and access to the original keys within the index. However, the special nature of genomic data and the specialized purpose of mapping mate-paired reads to a reference genome, can be exploited to create hash indices that are free from these limitations.
In SAWTooth, the hash key is the sequence tag, and the data to be retrieved is an exhaustive list of loci where the reads map in the reference genome. To demonstrate the feasibility of a cSBL approach to genome sequencing and calculate gains in using cSBL over traditional SBL methods, SAWTooth was applied to simulate human genome coverage using mate-paired data ranging from twenty-six bases (limit of traditional SBL) to forty bases (theoretical gain from cSBL implementation). A set of simulated mate-paired tags, each separated by a range of 300-700 bases, was created, ranging in size from 13 paired tags to 20 paired tags. A sufficient number of tags were computationally generated to simulate 10 × coverage. The tags were all generated from chromosome 1, mapped back to the entire genome, and calculations of chromosome 1 coverage were performed. Next, an analysis was performed of how many times each tag mapped to the genome. One of the more significant benefits gained by increasing tag length from 13 to 20 bases is that far fewer tags must be discarded because they do not map uniquely. At a tag length of 13 bases, only 57.2% of the tags are used, compared to 85.6% at a tag length of 20, thus effectively increasing throughput.
Address Goals
Primary: This project involves not only the bench sciences portion of working with biochemical processes and DNA, but also implementation of simulation data using an in-house developed software called the Sequencing Analysis Workbench Tool (SAWTooth). Antoine Ho was involved in the generation of simulation data and was a biologist consultant for the development of the SAWTooth package. The two accomplishments described above are in vastly different fields, one being a molecular biology advance and the other from computer science. Next-generation sequencing is inherently interdisciplinary and this project highlights the need for such collaboration to advance the field.
Secondary: The innovation of the cSBL variation extends and improves sequencing acquisition biochemistry, whereas the creation of SAWTooth improves the alignment of mate-paired tags to a reference genome. These are two key aspects of sequencing that needed to be improved, and make it more feasible for human genome sequencing. This in turn increases the effectiveness of categorizing Single Nucleotide Variations in an individual’s genome that may contribute to disease states. It is hoped that this knowledge may be used diagnostically, and then one day, therapeutically.


