

Highlight
The 1.9 structure of human Alpha-N-acetylgalactosaminidase: the molecular basis of Schindler and Kanzaki diseases
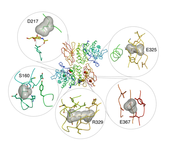
Figure 1: Schindler and Kanzaki disease mutations.
Achievement/Results
Nat Clark (IGERT trainee) and Scott Garman (IGERT advisor) of the University of Massachusetts Amherst have determined the three-dimensional crystal structure of human alpha-N-acetylgalactosamine (alpha-NAGAL) by x-ray crystallography. The lysosomal glycoprotein breaks down metabolic by-products in the cell and is defective in patients with the inherited metabolic disorders Schindler and Kanzaki diseases. The structure was solved by engineering out one of the wild type glycosylation sites and expressing the protein in insect cell culture. This research was recently published in the Journal of Molecular Biology (2009) and adopted an interdisciplinary approach drawing from the fields of molecular biology, microbiology, protein biochemistry, molecular modeling, enzymology, and glycobiology. The structure reveals the catalytic mechanism of the enzyme, explains the defects that lead to disease, and points to new directions for patient therapies.
The structure of human alpha-NAGAL has long been sought by researchers. Purified protein has been available since 1977, yet no structure has been determined despite considerable effort from multiple research groups. The investigators were able to determine the structure by judicious use of site directed mutagenesis combined with cutting edge protein expression technology. The structure represents one of the highest resolution human lysosomal structures, meaning that it will act as a model for a large family of lysosomal enzymes.
The human lysosomal enzyme alpha-NAGAL (E.C. 3.2.1.49) removes terminal alpha-GalNAc monosaccharides from glycolipids and glycoproteins (primarily O-linked sugars attached to serine and threonine residues). Deficiency in alpha-NAGAL leads to the lysosomal storage disorders Schindler disease and Kanzaki disease, first identified in 1987. In lysosomal storage disorders, loss of enzyme activity in a patient leads to the accumulation of substrate in the tissues, which ultimately leads to the development of clinical symptoms. In the lysosomal storage disorders Schindler and Kanzaki diseases, loss of functional alpha-NAGAL enzyme causes accumulation of glycolipids and glycopeptides, which ultimately results in neurologic and other pathologies.
In a recombinant insect cell expression system, the functional wild type glycoprotein was produced as well as mutants lacking each of the N-linked glycosylation sites. Enzymatic parameters of the wild type and mutant enzymes were measured, establishing the equivalence of the mutants. The structure of human alpha-NAGAL was determined to 1.9 Å resolution, revealing the mechanism of the enzyme. To understand the binding specificity of the human alpha-NAGAL enzyme, we determined crystallographic complexes with two catalytic products (the alpha-galactose and alpha-GalNAc monosaccharides) bound in the active site of the enzyme, as well as a complex with covalent intermediate bound. To better understand how individual defects in the alpha-NAGAL glycoprotein lead to Schindler and Kanzaki disease, the mutations that lead to disease were analyzed in light of the new three-dimensional structure of the glycoprotein. Overall, these results will lead to better understanding of the molecular basis of Schindler and Kanzaki diseases and will lead to better treatments for lysosomal storage diseases and protein folding diseases.
The metabolic disorders Schindler and Kanzaki diseases are caused by inherited mutations, leading to small changes in the structure of the alpha-NAGAL glycoprotein. The point mutations that cause disease include S160C, D217N, E325K, R329W, R329Q, and E367K. The structure of human alpha-NAGAL now reveals the molecular mechanism of Schindler and Kanzaki diseases (see Figure 1). The crystal structure reported here shows that Schindler and Kanzaki diseases are generally protein-folding diseases; thus, they may be treatable using not only enzyme replacement strategies (such as those used for Fabry and Gaucher patients), but also pharmacological chaperone approaches.
Address Goals
Our achievement primarily addresses a discovery goal. This work will impact understanding of lysosomal cell biology, and advance treatment options for patients with Schindler disease. This work opens the door for new avenues of study of lysosomal storage disorders. Additionally, our work supports research infrastructure. In order to make sufficient protein, new technology, the Wave Bioreactor, was employed. Through the ICE IGERT, we purchased this bioreactor, and have made it available to the campus community. It has expanded our University’s experimental and training capabilities, enabling cutting edge research. This bioreactor was used to teach large-scale cell culture techniques to an audience of graduate and undergraduate students across several disciplines, such as biology, psychology, chemical engineering, polymer science, chemistry, and biochemistry.
